Una malattia si definisce "rara" quando la sua prevalenza, intesa come il numero di caso presenti su una data popolazione, non supera una soglia stabilita. In UE la soglia è fissata allo 0,05 per cento della popolazione, non più di 1 caso ogni 2000 persone. Il numero di malattie rare conosciute e diagnosticate è di circa 10.000, ma è una cifra che cresce con l’avanzare della scienza e, in particolare, con i progressi della ricerca genetica. Stiamo dunque parlando non di pochi malati, ma di milioni di persone in Italia e circa 30 milioni in Europa. [Fonte: Eu Commission] Secondo la rete Orphanet Italia, nel nostro Paese i malati rari sono circa 2 milioni: nel 70% dei casi si tratta di pazienti in età pediatrica.
In base ai dati coordinati dal Registro Nazionale Malattie Rare dell'Istituto Superiore di Sanità (ISS), in Italia si stimano 20 casi di malattie rare ogni 10.000 abitanti e ogni anno sono circa 19.000 i nuovi casi segnalati dalle oltre 200 strutture sanitarie diffuse in tutta la penisola. Il 20% delle patologie coinvolge persone in età pediatrica (di età inferiore ai 14 anni). In questa popolazione di pazienti, le malattie rare che si manifestano con maggiore frequenza sono le malformazioni congenite (45%), le malattie delle ghiandole endocrine, della nutrizione o del metabolismo e i disturbi immunitari (20%). Per i pazienti in età adulta, invece, le malattie rare più frequenti appartengono al gruppo delle patologie del sistema nervoso e degli organi di senso (29%) o del sangue e degli organi ematopoietici (18%). [Fonte: ISS 2015]
Vista la mancanza di un’univoca definizione di malattia rara a livello internazionale, ci sono diverse liste di patologie: - National Organization for Rare Disorders (NORD) - Office of Rare Diseases - Orphanet (che propone una lista di circa 6.000 nomi di patologie rare, sinonimi compresi). In Italia, l’Istituto Superiore di Sanità ha individuato un elenco di malattie rare esenti-ticket. Alcune Regioni italiane hanno deliberato esenzioni per patologie ulteriori da quelle previste dal Decreto 279/2001.
Dott. Simone Ferrero (Torino): “Affinché si arrivi alla diagnosi, ematologo, reumatologo, immunologo e anatomo-patologo devono sospettare la malattia e interagire tra di loro”
Dispnea, stanchezza, dolori muscolari - a volte localizzati al torace, al dorso o alle articolazioni - tosse con espettorato di sangue e infezioni del tratto respiratorio. Questa sequenza di sintomi è riconducibile a un lungo elenco di ipotesi patologiche, lasciando pensare al medico di trovarsi di fronte a un tumore polmonare, oppure a una forma di leucemia o a una patologia infiammatoria. Ma potrebbe anche trattarsi di un disturbo raro, come quello scoperto settant’anni fa da Benjamin Castleman e che ancora porta il suo nome: la malattia di Castleman. Coloro che, al sentire questo nome, abbiano inarcato un sopracciglio assumendo un’aria interrogativa, possono trovare alcune spiegazioni sul sito dell’Associazione Malati Italiani Castleman (AMICa ODV) oppure guardare l’intervista-video di OMaR al vicepresidente, il dottor Simone Ferrero, ematologo presso l’Azienda Ospedaliero-Universitaria “Città della Salute e della Scienza” di Torino (clicca qui o sull’immagine dell’articolo per guardare il video).
Secondo uno studio, le linee guida emanate dalle istituzioni dovrebbero contenere indicazioni più precise sulla tipologia degli ingredienti utilizzati
Gli alimenti speciali a basso contenuto proteico (SLPF) sono cruciali per garantire un adeguato apporto calorico, migliorare l'aderenza alla dieta, prevenire il catabolismo e mantenere un buon controllo metabolico nei pazienti con malattie metaboliche ereditarie. Un recente studio pubblicato su JIM – Journal of Innate Metabolism ha analizzato la composizione degli SLPF in Italia e li ha confrontati con alimenti regolari (RF) simili.
L’Agenzia Europea per i Medicinali ha raccomandato la revoca dell’approvazione del farmaco. Le associazioni AISF, EpaC e AMAF fanno il punto della situazione
Lo scorso 28 giugno, il Comitato per i Medicinali per Uso Umano (CHMP) dell'Agenzia Europea per i Medicinali (EMA) ha emesso un parere negativo in merito all’acido obeticolico (nome commerciale Ocaliva), raccomandando alla Commissione Europea di revocare l'autorizzazione alla commercializzazione del farmaco. In Europa, l’acido obeticolico aveva ottenuto nel 2016 l’approvazione condizionata per il trattamento della colangite biliare primitiva (PBC), una malattia autoimmune del fegato, sulla base dei risultati di studi clinici in cui la molecola aveva dimostrato di ridurre i livelli ematici di fosfatasi alcalina (ALP) e bilirubina, due importanti marcatori di danno epatico. In linea con quanto previsto dalla procedura di approvazione condizionata, i reali benefici dell’acido obeticolico nei pazienti con PBC dovevano essere confermati in ulteriori sperimentazioni, ma i dati dello studio clinico 747-302 (COBALT), condotto a tale scopo, si sono rivelati deludenti e hanno indotto il CHMP ad emettere un parere sfavorevole in merito all’impiego del farmaco.
Con la Prof.ssa Maddalena Casale il punto sulle novità previste dalla Rete Nazionale Talassemia ed Emoglobinopatie
Il centro di Ematologia e Oncologia Pediatrica dell’Azienda Ospedaliera Universitaria Luigi Vanvitelli di Napoli afferente alla rete europea delle malattie ematologiche rare (ERN Eurobloodnet) è un centro di riferimento per le emoglobinopatie, pediatriche e dell’adulto. Il centro, diretto dal Prof. Silverio Perrotta, dispone di un team dedicato alle emoglobinopatie, supportato da una rete di specialisti esperti, dall’endocrinologo, al cardiologo, all’ortopedico fisiatra, al radiologo, al neuropsichiatra ed altri specialisti. “Seguiamo tutte le forme di emoglobinopatie – spiega la Prof.ssa Maddalena Casale, ricercatrice del centro – dalle sindromi talassemiche, alle drepanocitosi, alle forme da emoglobine anomale instabili ad alta e bassa affinità per l’ossigeno.
Sono quattro i Paesi nel continente europeo in cui il trattamento è ora disponibile. I pazienti italiani sono invece ancora in attesa
Milano – Etranacogene dezaparvovec,terapia genica per l’emofilia B prodotta da CSL Behring, continua ad ampliare la propria diffusione in Europa: recentemente il farmaco è stato infatti rimborsato in Danimarca e Regno Unito, dopo le autorizzazioni ricevute in Francia e Austria. Si tratta della la prima terapia genica approvata in Europa per il trattamento dei pazienti eleggibili con l’ emofilia B, una malattia emorragica ereditaria causata dalla mancanza del fattore IX (una proteina necessaria per la coagulazione del sangue).
Procedono le sperimentazioni del farmaco nella colangite biliare primitiva e nella colangite sclerosante primitiva
L’azienda Mirum Pharmaceuticals ha recentemente annunciato i positivi risultati ad interim di due studi clinici di Fase IIb attualmente in corso per valutare la sicurezza e l’efficacia del farmaco orale volixibat, un inibitore del trasportatore ileale degli acidi biliari, per il trattamento di pazienti affetti da colangite biliare primitiva (PBC) o da colangite sclerosante primitiva (PSC), due malattie autoimmuni del fegato.
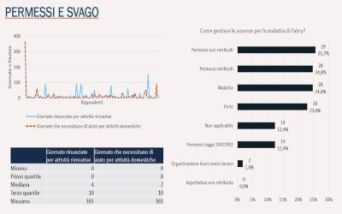
È quanto emerge dalla survey nell’ambito del progetto Caring Fabry, condotta da AIAF in collaborazione con Helaglobe
Sono circa 20 le giornate che, in media, una persona con malattia di Fabry perde ogni anno per recarsi in ospedale e sottoporsi a visite e terapie: è quanto emerge da un’indagine realizzata dall’Associazione Italiana Anderson-Fabry (AIAF-APS), un dato già rilevato nel 2018 e riconfermato attraverso una survey più recente, nell’ambito del progetto “Caring Fabry”, realizzato in collaborazione con la Società Helaglobe Srl. Obiettivo: colmare la disparità tra bisogno di assistenza e attuale presa in carico, disegnando il modello ideale di presa in carico del paziente con malattia di Anderson-Fabry. In questo modo potranno essere ottimizzati i tempi e potrà essere migliorato il funzionamento degli attuali centri di cura.



















Seguici sui Social